


{kind=link}

RNA can adopt many configurations in the cell (artist’s illustration).Credit: Christoph Burgsted/Science Photo Library/Getty
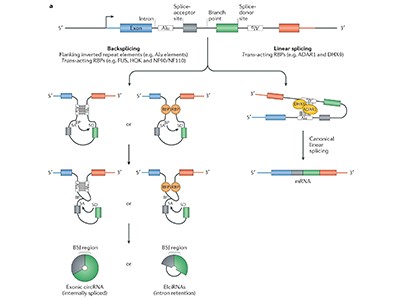
Over the past few decades, RNA’s place in biology has transformed from being a mere intermediate between DNA and protein to a fascinating molecule with diverse activities that go well beyond simple transcription of genetic information. Many of these RNAs fold up like molecular origami, but one of their most puzzling configurations is circular: molecules in which an unusual version of the standard RNA-splicing process folds the strand back on itself, creating a loop.
Once thought to be artefacts of splicing gone wrong, circular RNAs (circRNAs) are now known to be widespread across the tree of life. They’ve been implicated in conditions including cancer, cardiovascular disease and Alzheimer’s disease, and offer exciting possibilities as both therapeutic agents and biomarkers.
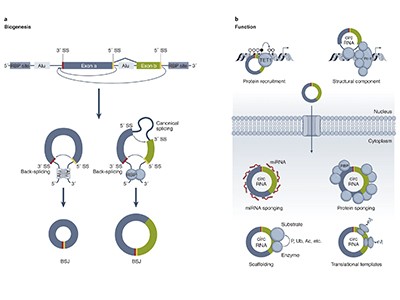
Even so, scientists are still working out what these molecules do. Some of the first circRNAs to be studied mop up small, non-coding RNAs called microRNAs to prevent them from binding to messenger RNAs and dampening protein production. But others might interact with various proteins or the enzyme RNA polymerase to regulate transcription and protein expression, or even be translated themselves (see ‘Potential functions of circular RNA’).

Source: V. M. Conn et al. Nature Rev. Cancer 24, 597–613 (2024).
“There are really very interesting things to be found out about circular RNAs,” says Nikolaus Rajewsky, a systems biologist at the Max Delbrück Center for Molecular Medicine in Berlin. “Largely, it’s terra incognita, and that’s very exciting.”
Not to mention challenging. That’s in part because circRNAs are rare — they make up about 0.1% of non-ribosomal RNA sequences, by one measurement1 — and also because they’re essentially identical to linear RNAs transcribed from the same part of the genome. Their only distinguishing feature is where they’re joined together to form a circle, that is, where the end of one RNA segment links up with a segment from earlier in the DNA code. This is called the back-splice junction. It’s difficult to analyse or generate pure circular forms without the linear ones interfering, and specialists warn that the scientific literature is littered with spurious findings that link a particular circRNA with a specific microRNA without iron-clad evidence. “You really need to do a thousand controls,” says Rajewsky.
Researchers who are new to the field can follow published best practices2,3. But the ideal approach is to consult with circRNA veterans before starting a project, suggests Grace Chen, an RNA biologist at Yale University School of Medicine in New Haven, Connecticut. “Talk to us early, and talk to us often,” she says.
That said, researchers are adapting a variety of methods from standard RNA biology to identify and investigate these exciting molecules, and the circRNA-specific toolbox is growing. “I think people should do the hard work to get into this field if this is something that they’re interested in,” says Chen. “There’s a lot to be discovered here.”
Loopy molecules
Step one is to identify circRNAs of interest. There are multiple databases for this, but many are uncurated, incomplete and riddled with unvalidated listings, cautions Jo Vandesompele, a cancer-genomics researcher at Ghent University in Belgium who reviewed4 circRNA databases with his colleagues in 2020. The naming system is a jungle, he says, with many names for the same molecule, and the database landscape a full-scale nightmare. If he had to pick one database, he says, it would be circAtlas from the Beijing Institutes of Life Science in China. That’s because circAtlas requires any listed circRNAs to be identified by two tools, each with at least two counts of the back-splice junction.

Why rings of RNA could be the next blockbuster drug
Sequencing is the most common approach to find new circRNAs, but these loopy molecules are often missing from standard RNA libraries. Scientists usually build these libraries by targeting the poly(A) tail attached to mRNAs — a feature that circRNAs lack. “You need to be specifically looking for them,” says Jeremy Wilusz, an RNA biologist at Baylor College of Medicine in Houston, Texas.
More specifically, circle hunters must seek out that back-splice junction. It is not a single signature sequence, but any place where the 3′ end of one coding sequence, or exon, connects to the 5′ end of an exon that is normally upstream in the linear sequence — and non-coding sequences called introns can also get in on the act.
The most common option is to create total RNA libraries, says Vandesompele: it’s easier and less expensive than making circRNA-specific libraries, and means scientists can look for multiple RNA forms, not just circles. First, however, they have to get rid of ribosomal RNA, a structural component of ribosomes that represents the vast majority of RNAs in cells.
Given the low abundance of most circRNAs, getting enough material to sequence is part of the challenge, says Prisca Obi, an immunobiologist and newly minted PhD graduate in Chen’s laboratory at Yale University. The cost of commercial kits to eliminate ribosomal RNA would quickly add up before scientists obtained much circRNA. So, Chen and her colleagues developed an inexpensive, do-it-yourself protocol5. They add DNA probes containing sequences that match ribosomal RNA, then use the enzyme RNase H to destroy the resulting RNA–DNA hybrid molecules. After a few further clean-up steps, “you should have RNA that’s mostly depleted of ribosomal RNA”, says Obi.
The alternative is to strip away most of the non-circular RNAs, too. This requires the enzyme RNase R, which attacks linear RNAs at their ends. But it’s important to titrate the reaction conditions carefully, Vandesompele warns: too much enzyme will destroy the circles, and too little will spare some of the linear pieces. Also, certain RNA structures, such as G-quadruplexes, histone mRNAs and small nuclear RNAs, can foil the enzyme. Swapping the potassium in the reaction buffer for lithium destabilizes these structures and helps to improve the degradation of linear RNAs, Wilusz and a colleague have reported6.
Telltale junctions
Once researchers have sequenced the RNAs — short-read methods such as those developed by US biotechnology firm Illumina are generally fine, says Vandesompele — the next step is to search those sequences for the telltale back-splice junctions . But the algorithms available for doing so vary widely in sensitivity.
In a 2023 comparison of 16 tools conducted by Vandesompele and his colleagues, individual algorithms identified anywhere from 1,372 to 58,032 circRNAs from the same cell line7. Vandesompele recommends researchers select at least two tools that have low false-positive rates; his lab has landed on CirComPara2 and circtools, but others might have different needs.
Best practice standards for circular RNA research
None of these algorithms is perfect, Rajewsky notes: “In our experience, about 80% of these things are real.” Unexpected splice forms or amplification issues can create spurious results.
Another catch is that most search algorithms rely on matching RNA sequences to a reference genome, says Julia Salzman, a computational biologist at Stanford University in California. If the circRNA sequence is missing from the reference genome — because, say, you’re searching for a viral circRNA but the reference sequence is human — it will never be found, creating a false-negative result. And because near-homologous sequences are found throughout the genome, attempts to match the sequences can also create false positives.
Salzman and her colleagues developed an alternative approach called SPLASH2. The software compares two sets of sequences without relying on a reference genome, on the basis of counts of short genetic segments called k-mers. Salzman recommends comparing a sample containing both circles and linear RNAs with one treated with RNase R to reduce the linear component — differences between the two samples will point to circRNAs. In one specificity test, 92% of potential circRNAs identified by SPLASH2 were known or likely circles8.
Circular validation
To quantify recognized circRNAs, scientists can use microarray chips studded with nucleic-acid probes for known circRNA back-splice junctions. Scientists can wash their samples over the chip to detect when a sequence in the sample matches its partner on the array.
Arraystar, a biotechnology company focused on non-coding RNAs in Rockville, Maryland, has designed circRNA microarrays targeting the known circles for human, mouse and rat samples, and researchers have used them, among other things, to identify a circRNA in mouse blood stem cells that helped the cells to avoid exhaustion9. A key caveat: “If it’s not there on the array, it could be super-interesting, and you’ll never find it,” says Wilusz.
Yanggu Shi, a senior scientist at Arraystar, says that about 70% of microarray predictions hold up. So, the next step for any detection approach is to validate that the circRNAs are present and genuinely circular — typically by using quantitative PCR. Researchers treat part of a sample with RNase R to degrade linear RNAs and leave part untreated, and amplify both samples; any real circles should withstand the enzyme treatment, whereas linear counterparts would be diminished.
That said, the back-splice junction represents just one part of the RNA: there could be several circles, each with different complements of exons or even introns sharing the same junction. That’s why Wilusz’s validation strategy includes non-PCR approaches, such as Northern blotting, in which researchers separate RNA molecules by size and then use sequence-specific probes to detect molecules of interest. That way, they can not only detect specific RNAs, but can also determine how many molecules of different sizes contain those sequences. Long-read sequencing, although expensive, is another option. “Only then can you have an unambiguous identification,” says Vandesompele.
The biogenesis, biology and characterization of circular RNAs
The abundance of a circRNA can provide an important clue to its function, says Vanessa Conn, a molecular biologist at Flinders University College of Medicine and Public Health in Bedford Park, Australia. For example, if scientists predict that a circRNA is mopping up microRNAs, then the circRNA should be sufficiently abundant to catch most of the microRNAs in a cell. But even a rare circRNA might be able to influence a gene at the DNA level, because there are only two copies of it in the genome. For example, Conn, together with her husband Simon Conn, a molecular cancer biologist who leads a lab at Flinders University, discovered one low-level circRNA that interacts with DNA to drive genetic translocations associated with leukaemia10.
Quantifying circRNAs can be tricky, the Flinders researchers say. That’s because small circRNAs in a sample will be amplified more quickly than are large ones, making the small molecules seem more abundant than they are. Conn and his group developed a method they call SplintQuant to get around that problem11. Once scientists have identified a circle of interest, they can design DNA probes to match either side of its back-splice junction. Then they use an enzyme to link up any pairs of probes that have found a circle together, and use quantitative PCR to count the ligated molecules. But what about comparing circRNA sequences and quantities between labs and protocols? The Conns propose a solution here, too, in the form of synthetic circRNAs called SynCRS (pronounced sinkers) that researchers can spike into their samples before library production12. Using known quantities of SynCRS allows scientists to normalize results across labs.
Muddied waters
From there, scientists can finally move on to the most interesting challenge: function. Key approaches include knocking down or overexpressing the circles, and again, similarities to other RNA species muddy the waters.
A technique called RNA interference is one common approach, because it can be targeted to the back-splice junction, says Guillermo Aquino-Jarquin, a researcher in medical sciences at the Federico Gómez Children’s Hospital of Mexico in Mexico City. For example, by targeting the cancer-linked circRNA circAGO2 in mice, using small hairpin RNAs, researchers demonstrated the gene’s role in promoting tumour generation and aggressiveness13. Alternatively, if researchers suspect that specific circle sequences have binding partners among the nucleic acids or proteins in a cell, they can design interfering RNAs to bind to and suppress those sites specifically.
The alternative is to use the CRISPR–Cas system to target and obliterate circles of interest. With Cas9, a DNA-targeting enzyme, researchers can damage a circRNA gene itself, destroying its ability to make the molecules. Rajewsky and colleagues used this strategy to make mice deficient in the circRNA Cdr1as, which interacts with the microRNA miR-7. Without Cdr1as, cells contained less miR-7, suggesting that the circRNA controls the microRNA’s stability or perhaps its transport14.
That said, linear RNA production can also be affected by alterations to the genome. Alternatively, researchers can use the enzyme Cas13 to target the back-splice junction or other binding sites at the RNA level15. “You knock down the circRNA, but you don’t touch the genome,” says Aquino-Jarquin. Knockdowns of this type are about 80–90% effective, he estimates, compared with 50–60% effectiveness for RNA interference. Cas enzymes can also create off-target effects, however.

NatureTech hub
Thus, as with circularity itself, validation and controls remain key, says Wilusz. For example, researchers who think that a circRNA might be sponging up microRNAs might eliminate the circle itself, as well as mutating likely microRNA binding sites and looking for similar results. And, if eliminating a circle produces a phenotype, adding that circle back in should reverse it. That’s where overexpression comes in. This is doable, but impossible to achieve perfectly, says Rajewsky. “Nobody can produce, as far as I know, 100% clean, circular RNAs,” he says. “You will also introduce some other RNAs.”
Researchers can generate circRNAs synthetically or make them in vivo using plasmids that are designed to use the cell’s own splicing machinery. The latter approach “would, ideally, most faithfully recapitulate the cell making it”, says Obi. One classic approach, the permuted intron–exon (PIE) strategy, involves rearranging exons and introns into configurations that promote automatic release of a circularized product16. Plasmids that generate circRNAs are available from the US non-profit plasmid repository Addgene.
For the best control over circRNA purity, synthetic benchtop methods are preferable, Obi says. She and Chen like to generate linear RNA precursors, then link them together with an enzyme called ligase. To minimize undesirable side products, researchers can add RNase R to degrade unwanted linear RNAs, and use separation techniques such as gel electrophoresis or liquid chromatography to purify the desired species.
But as always, Rajewsky warns, there are caveats. With synthetic circles, “whatever you’re putting in is artificial, and might not relate at all to the biology that’s happening in the cells.”
So when it comes to circRNAs, the key is to test hypotheses with multiple methods, he says. For example, Rajewsky and his colleagues recently reported that the circRNA Cdr1as interacts with miR-7 to regulate the release of the neurotransmitter glutamate from neurons17. They used a variety of techniques: mammal primary neuron culture, chemical and electrical stimulation, single-cell RNA imaging and microRNA knockouts — to name a few.
Is that overkill? Not when it comes to circRNAs, says Rajewsky. “It is necessary to really say something substantial about these crazy molecules.”