


{kind=link}
For most of her life, Genesis Jones’s daily routine revolved around her illness, the painful blood disorder known as sickle-cell disease. Each time she left the house, she ran through a mental checklist: did she have her pain medications? What was her energy level? Would she be able to make it through the day?
Jones, who lives in Grand Prairie, Texas, longed to be free from the constant threat of a severe pain crisis or a stroke. So, in 2020, she opted for the only potential cure at the time: a transplant of blood stem cells from a donor without the disease. She spent six months preparing for the procedure, including weeks of radiation, chemotherapy and a lengthy hospital stay to destroy her own blood stem cells and make room for cells donated by her mother. The procedure cured her sickle-cell disease, and she is grateful.
“It’s great to have that pain and anxiety lifted,” she says. “But it’s not the end of the story.”
Less than one month after her transplant, Jones learnt that she had cancer, a known risk of the treatment. Three more rounds of chemotherapy and other treatments drove her cancer into remission, but she still struggles with chronic pain in her back and legs caused by decades of tissue and nerve damage from sickle-cell disease. And she worries that signs of mild cardiac inflammation mean that her new stem cells are making immune cells that are attacking her heart.
Then there are the mental-health consequences. Jones’s treatment alienated her from the sickle-cell-disease support networks she had come to rely on, even though she was still wrestling with the consequences of the condition. “I feel like I’m in-between,” she says.

Hope, despair and CRISPR — the race to save one woman’s life
That in-betweenness extended to her medical care: because blood tests no longer indicated she had sickle-cell disease, physicians sometimes turned her away when she sought help for her pain. “They say, ‘Well, why are you here?’” she says. “Basically, you have to suffer alone at home.”
Over the past decade, stem-cell transplants and gene therapies for treating sickle-cell disease have blossomed, offering fresh hope to people with severe illness. Researchers have improved protocols for stem-cell transplants, and the US Food and Drug Administration (FDA) approved two genetic treatments last year — including the first CRISPR-based genome-editing therapy the agency has ever authorized.
But the success stories that make headlines belie the uncertainties and struggles that still surround the treatments. Just as surviving cancer can leave a mark on someone’s mental, financial and physical health, so too can these potentially curative cell and gene therapies. But, unlike for people with cancer, awareness and support are often lacking for those who have been ‘cured’ of sickle-cell disease, a condition often affecting people from communities that already face discrimination and inequality in access to health care. “There is a lack of education among health-care providers on how these patients should be managed,” says Akshay Sharma, a specialist in bone-marrow transplants at St. Jude Children’s Research Hospital in Memphis, Tennessee.
Similar concerns weigh on people who have received gene therapies for other conditions, and that number is growing rapidly. About 30 gene therapies have been approved in the United States and more than 600 others are undergoing clinical testing around the world. Clinicians and patients are now wrestling with how to support people after their treatment. “Gene therapy is going to change the entire landscape,” says Anirban Basu, a health-care economist at the University of Washington in Seattle. “This is coming.”
Progress in gene therapies
Sickle-cell disease is caused by a mutation in the gene encoding a component of haemoglobin, the oxygen-carrying protein complex found in red blood cells. The dysfunctional haemoglobin distorts the cells, which are usually round, into a sickle or crescent shape. Those misshapen cells can clog blood vessels, which deprives tissues of oxygen, causing severe bouts of overwhelming pain, called pain crises.
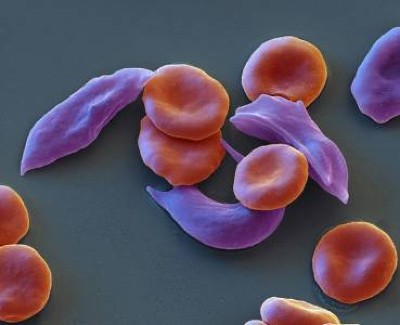
UK first to approve CRISPR treatment for diseases: what you need to know
Clogged vessels can also cause strokes and damage organs, particularly the liver, heart and kidneys, over time. In the United States, people with sickle-cell disease have an average life expectancy that is some 20 years shorter than people without it1.
Sickle-cell disease is often treated with hydroxyurea, a drug that can reduce the frequency of pain crises. For decades, the only treatment that could eradicate the disease was a blood-stem-cell transplant from a matched donor.
But globally, few people with sickle-cell disease have received transplants, because of the difficulty of finding suitable donors, the expense and risks entailed in the procedure and the effects of systemic racism. Biased and discriminatory medical care is often a fact of life for people of sub-Saharan African or Indian ancestry, who are more likely to have the sickle-cell mutation owing to the prevalence of malaria in those regions. The mutation is beneficial because the parasite that causes malaria is less able to grow in crescent-shaped blood cells.

Jones appeared in a national advertising campaign to support blood-stem-cell donation.Credit: Nitashia Johnson for Nature
Some barriers are falling, however. Steady work to improve transplant protocols has boosted success rates and lowered toxicity, says Michael DeBaun, who specializes in treating sickle-cell disease at Vanderbilt University Medical Center in Nashville, Tennessee. In November, DeBaun and his colleagues showed that stem cells from only partially matched donors — such as a parent — could be transplanted successfully in people with sickle-cell disease who received a lower-than-usual dose of radiation and chemotherapy before the procedure2.
At the same time, gene-targeting therapies have also progressed. One therapy, called Lyfgenia (lovotibeglogene autotemcel), provides a working version of the gene that is affected in people with the disorder. The other therapy, called Casgevy (exagamglogene autotemcel), uses CRISPR–Cas9 genome editing to reactivate a form of haemoglobin that is normally inactivated soon after birth. This fetal haemoglobin helps to compensate for the dysfunctional version.
Neither of these gene therapies corrects the mutation that causes sickle-cell disease, and neither can be considered a cure. But both drastically reduce the incidence of acute pain crises in people who receive treatment, and there is hope that those benefits will be long-lasting. In December 2023, biotechnology company Bluebird Bio in Somerville, Massachusetts, which makes Lyfgenia, reported that the therapy’s benefits were durable for at least five years3. The company continues to follow participants in its clinical trials to see how long the effects last.
Treatment at a price
All of the treatments are expensive: a stem-cell transplant costs between about US$100,000 and $400,000 and the list price of Casgevy, which was developed by Vertex Pharmaceuticals in Boston, Massachusetts, and CRISPR Therapeutics in Zug, Switzerland, is $2.2 million. Such price tags mean that these therapies are out of reach for many people with sickle-cell disease around the world. In the United States, an effort is under way to make cell and gene therapies available to people who receive publicly funded health care. The programme is slated to go into effect in 2025, but it is not yet clear how many states will take part in it. In the United Kingdom, the National Institute for Health Care and Excellence determined in March that Casgevy is not cost-effective for treating sickle-cell disease and asked the developers for further data. As a result, Casgevy is not yet available for treating sickle-cell disease through the country’s public health-care system.
Both stem-cell transplants and gene therapy require chemotherapy before the treatment. Chemotherapy is time-consuming and risky. Women who go through it are often left infertile. They can opt to have their eggs frozen before treatment, but in the United States, the harvesting, freezing and storage of eggs typically costs more than $10,000.

‘It’s a vote for hope’: first gene therapy for muscular dystrophy nears approval, but will it work?
That price was too high for Teonna Woolford, who once dreamed of having six children — before she received a stem-cell transplant at the age of 19. By then, her sickle-cell disease was landing her in hospital about every other week. She’d had both hips replaced because of bone damage caused by impaired blood flow and her liver was failing. Her doctors urged her to consider a stem-cell transplant.
Woolford, who lives in a suburb of Baltimore, Maryland, reached out to a few cancer charities that offer assistance to try and retain fertility for women undergoing chemotherapy. “I don’t have cancer, but I’m getting chemo and radiation,” she explained. “Maybe you can squeeze me in?” The answer was no.
In addition to infertility, the chemotherapy can increase the risk of bone degeneration, which is already elevated in people with sickle-cell disease. The chemotherapy regimen can also raise the risk of cancer. DeBaun is hopeful that new transplant protocols that use lower doses of chemotherapy will reduce the chances of these side effects.
As for gene therapies, researchers say it is important to follow recipients for years after their treatment to monitor for any increase in cancer rates, either from the chemotherapy regimen or the gene therapy itself. Lyfgenia carries a warning about cancer risk because two clinical-trial participants later developed leukaemia. Long-term data will be crucial to determine whether either Lyfgenia or Casgevy truly pose a cancer risk, or whether such cases are associated with the underlying sickle-cell trait, which might also predispose people to blood cancers, says Mark Walters, a paediatric haematologist and oncologist at the University of California, San Francisco.
For now, it’s difficult to tease these risks apart because the clinical trials have been relatively small, and could not include a control group, says James LaBelle, a paediatric oncologist at the University of Chicago Medicine in Illinois. Participants in clinical trials were also carefully selected, he notes, and lacked some of the organ damage or history of stroke that might be present in people with severe sickle-cell disease, making it hard to know how well the gene therapies will work — and how safe they will be — in that population.
Looking to the long term
As the therapies expand from clinical trials to hospitals, there is an opportunity to collect data that would address these questions, LaBelle says. The FDA has suggested that gene-therapy manufacturers should collect data from the recipients of their products for up to 15 years after treatment. Researchers outside those companies are also setting up a registry to track recipients of sickle-cell gene therapies, he adds.
But a lingering question is how to capture those data in the first place. Cell- and gene-therapy recipients often remain in the care of their transplant team for some time after their treatment. But they eventually return to their regular health-care providers, who might not know the full medical history of their patients, or the testing and data reporting expectations for someone who has received a gene therapy.

In search of a community, Jones formed a support group with others who have a copy of the sickle-cell-disease mutation.Credit: Nitashia Johnson for Nature
Then there is the difficulty of getting people with sickle-cell disease, some of whom are traumatized by hospitals and medical procedures, to come back every year for a follow-up examination, says Walters. “Some of the recipients just feel like, ‘I’m done with sickle-cell disease. The last thing I want to do is give you blood samples’,” he says. “I don’t know how we’re going to motivate participation.”
Long-term follow-up is a struggle for the wider gene-therapy field, too. Jack Grehan, a videographer in Manchester, UK, was treated for a blood-clotting disorder called haemophilia A more than five years ago. For the first few years after receiving the therapy, he returned to the hospital for follow-up exams each time he was asked to come. But now he lives two hours away from the treatment centre and is juggling childcare and a new job. He has not had a single bleeding episode since receiving the treatment, and he stopped going to check-ups two years ago. “I’ll happily go back and give them more once my life balances back out,” he says.
But in the years after Grehan was treated in a clinical trial, researchers found that the effects of the gene therapy he received can wane over time. As a result, physicians often urge recipients to have their blood-clotting proteins checked routinely. When first approached by Nature in March, Grehan was unaware of that concern.
Improve support for chronic issues
And some people will require continued care. Many with sickle-cell disease who receive transplants find fast relief from crippling, acute pain crises, but the chronic pain from decades of damage to organs and nerves can take much longer to address. “It can be such a source of disappointment for people because it doesn’t go away magically in three months,” says Walters. “The pain-sensation network in sickle cell is really disordered.”
Jones’s story of being refused care for her pain after her transplant echoes what several physicians and sickle-cell-disease advocates have told Nature. Stem-cell transplants or gene therapy can make the blood seem remarkably normal in recipients, says Sharma. “But they truly still have pain,” he says, “and this is something that emergency-room physicians or other clinicians are unable to grapple with.”
Gene therapy’s comeback: how scientists are trying to make it safer
Over time, many people will find relief even from chronic pain through therapy and pain-management techniques, without having to rely on the opioids they might have been prescribed before the transplant, says Lakshmanan Krishnamurti, a paediatric haematologist and oncologist at Yale School of Medicine in New Haven, Connecticut. But it can take hard work to get there, and health-care plans often do not cover the cost of that journey. It’s especially difficult for people from disadvantaged communities. “You do gene therapy and then throw these people back in the same environment. Why should I be surprised they don’t have access to pain management?” he says.
And there are mental-health challenges to navigate, too. After noticing that many transplant recipients seemed to struggle after their procedures, Elisabeth Dovern, a haematologist at Amsterdam University Medical Center, and her colleagues conducted a survey of health challenges that arise after a transplant.
The team found that most transplant recipients reported positive changes, including improvements in physical, mental and social health. But even among those who were happy with the outcome, some people struggled. Chronic pain and bone degeneration remained a problem for some recipients, as did feelings of isolation4. A lifetime of illness had made the hospital a source of trauma for some. They faced symptoms of post-traumatic stress disorder now that the ordeal was largely over. For others, the hospital was a second home, and they mourned the loss of that community when their health improved, says Dovern.

The sickle-cell drug boon
Many recipients of cell or gene therapy feel like they need to fill the space left by the condition they had spent a lifetime fighting. “The disease becomes the narrative,” says Krishnamurti. “It’s very difficult to change the narrative of a life.”
Dovern expects her findings for transplant recipients to translate to the experiences of those who receive gene therapy for sickle-cell disease. Indeed, recipients of other forms of gene therapy have wrestled with similar problems. Dovern and her colleagues now assign recipients of stem-cell transplants a psychiatric counsellor who meets with them before and after the procedure to help buffer the shock of the transition to a new life. Krishnamurti spends months preparing people not only for the procedure, but also for the turmoil that can come afterwards. “You describe yourself as a sickle-cell warrior? Your training to become a sickle-cell veteran starts now,” he tells them.
Late last year, when many sickle-cell-disease advocates were celebrating the FDA approvals of gene therapies, Woolford decided to start a support group for people who received transplants or gene therapy. The group has about 25 members. Jones has also formed a support group that is about ten members strong. “I needed to feel like I still had support within the sickle-cell community,” she says.
The members of Jones’s support group share stories and experiences, sifting out the misinformation they encounter along the way. In a recent virtual gathering, the group marvelled over Supacell, a drama series on streaming service Netflix in which a group of Black Londoners with sickle-cell disease in their families develop superpowers. They groaned in sympathy when one member recalled their struggles to get physicians to take their condition seriously.
Jones says that she is still trying to work out how best to make her voice heard by health-care providers unaccustomed to treating people with sickle-cell disease after a transplant. “Long-term care is often overlooked,” she says. “It does take a while for the body to catch up — and for the mind to get there, too.”